Mark Gorelik,1 Sharon A. Chung,2![]() Kaveh Ardalan,3
Kaveh Ardalan,3![]() Bryce A. Binstadt,4 Kevin Friedman,5 Kristen Hayward,6 Lisa F. Imundo,1 Sivia K. Lapidus,7
Bryce A. Binstadt,4 Kevin Friedman,5 Kristen Hayward,6 Lisa F. Imundo,1 Sivia K. Lapidus,7![]() Susan Kim,2
Susan Kim,2![]() Mary Beth Son,5
Mary Beth Son,5![]() Sangeeta Sule,8
Sangeeta Sule,8![]() Adriana H. Tremoulet,9 Heather Van Mater,3 Cagri Yildirim-Toruner,10 Carol A. Langford,11 Mehrdad Maz,12
Adriana H. Tremoulet,9 Heather Van Mater,3 Cagri Yildirim-Toruner,10 Carol A. Langford,11 Mehrdad Maz,12![]() Andy Abril,13 Gordon Guyatt,14 Amy M. Archer,15 Doyt L. Conn,16
Andy Abril,13 Gordon Guyatt,14 Amy M. Archer,15 Doyt L. Conn,16![]() Kathy A. Full,17 Peter C. Grayson,18
Kathy A. Full,17 Peter C. Grayson,18![]() Maria F. Ibarra,19 Peter A. Merkel,20 Rennie L. Rhee,20
Maria F. Ibarra,19 Peter A. Merkel,20 Rennie L. Rhee,20![]() Philip Seo,21 John H. Stone,22
Philip Seo,21 John H. Stone,22![]() Robert P. Sundel,5 Omar I. Vitobaldi,23 Ann Warner,24 Kevin Byram,25 Anisha B. Dua,15
Robert P. Sundel,5 Omar I. Vitobaldi,23 Ann Warner,24 Kevin Byram,25 Anisha B. Dua,15![]() Nedaa Husainat,26
Nedaa Husainat,26![]() Karen E. James,27 Mohamad Kalot,28 Yih Chang Lin,29 Jason M. Springer,25 Marat Turgunbaev,30 Alexandra Villa-Forte,11 Amy S. Turner,30
Karen E. James,27 Mohamad Kalot,28 Yih Chang Lin,29 Jason M. Springer,25 Marat Turgunbaev,30 Alexandra Villa-Forte,11 Amy S. Turner,30![]() and Reem A. Mustafa12
and Reem A. Mustafa12![]()
Guidelines and recommendations developed and/or endorsed by the American College of Rheumatology (ACR) are intended to provide guidance for particular patterns of practice and not to dictate the care of a particular patient. The ACR considers adherence to the recommendations within this guideline to be voluntary, with the ultimate determination regarding their application to be made by the physician in light of each patient’s individual circumstances. Guidelines and recommendations are intended to promote beneficial or desirable outcomes but cannot guarantee any specific outcome. Guidelines and recommendations developed and endorsed by the ACR are subject to periodic revision as warranted by the evolution of medical knowledge, technology, and practice. ACR recommendations are not intended to dictate payment or insurance decisions, and drug formularies or other third-party analyses that cite ACR guidelines should state this. These recommendations cannot adequately convey all uncertainties and nuances of patient care.
The American College of Rheumatology is an independent, professional, medical and scientific society that does not guarantee, warrant, or endorse any commercial product or service.
Objective. To provide evidence-based recommendations and expert guidance for the management of Kawasaki disease (KD), focusing on clinical scenarios more commonly addressed by rheumatologists.
Methods. Sixteen clinical questions regarding diagnostic testing, treatment, and management of KD were developed in the Patient/Population, Intervention, Comparison, and Outcomes (PICO) question format. Systematic literature reviews were conducted for each PICO question. We used the Grading of Recommendations, Assessment, Development and Evaluation method to assess the quality of evidence and formulate recommendations. Each recommendation required consensus from at least 70% of the Voting Panel.
Results. We present 1 good practice statement, 11 recommendations, and 1 ungraded position statement to guide the management of KD and clinical scenarios of suspected KD. These recommendations for KD are focused on situations in which input from rheumatologists may be requested by other managing specialists, such as in cases of treatment-refractory, severe, or complicated KD. The good practice statement affirms that all patients with KD should receive initial treatment with intravenous immunoglobulin (IVIG). In addition, we developed 7 strong and 4 conditional recommendations for the management of KD or suspected KD. Strong recommendations include prompt treatment of incomplete KD, treatment with aspirin, and obtaining an echocardiogram in the setting of unexplained macrophage activation syndrome or shock. Conditional recommendations include use of IVIG with other adjuvant agents for patients with KD and high-risk features of IVIG resistance and/or coronary artery aneurysms. These recommendations endorse minimizing risk to the patient by using established therapy promptly at disease onset and identifying situations in which adjunctive therapy may be warranted.
Conclusion. These recommendations provide guidance regarding diagnostic strategies, use of pharmacologic agents, and use of echocardiography in patients with suspected or confirmed KD.
Kawasaki disease (KD) is a medium vessel vasculitis as presented in the Chapel Hill Consensus Conference nomenclature system (1). It is 1 of 2 primary common vasculitis syndromes that predominantly affect children (the other being IgA vasculitis [Henoch-Schönlein]). KD is seen with relative frequency in young children, at an incidence rate of 25–50 cases per 100,000 persons per year in the US, similar to that of type 1 diabetes (2,3). It typically presents in children <5 years of age as an acute, self-limited febrile disease and is characterized by a combination of several characteristic clinical signs, which include polymorphic rash, nonpurulent conjunctival injection, oropharyngeal and lip mucositis, tongue papillitis, erythema and edema of the hands and feet, as well as unilateral cervical lymphadenopathy.
From a histopathologic perspective, KD is characterized by aggressive neutrophil-mediated panmural vascular necrosis without granuloma formation, followed by intimal hyperplasia and subacute lymphocytic inflammation in advanced disease. It primarily affects the coronary arteries but can also affect medium-caliber arteries throughout the body (4). Therefore, it shares some features with polyarteritis nodosa, and prior to the recognition of KD as a distinct entity, infants with fatal fulminant forms of KD were considered to have “infantile polyarteritis nodosa” (5).
When treated appropriately, KD is associated with a low mortality rate (~0.08% case fatality rate in the largest reported series) (6). However, in the developed world, it is the most common cause of acquired cardiac disease in childhood, with 25% of untreated patients and 5% of treated patients developing coronary artery aneurysms. Among infants <6 months of age, the risk of a coronary artery aneurysm is 50%, even in KD patients who received treatment with intravenous immunoglobulin (IVIG) within the first 10 days of illness (7).
Given increasing options available to treat systemic vasculitis, the American College of Rheumatology (ACR) and Vasculitis Foundation supported the development of guidelines for the management of large, medium, and small vessel vasculitis. This guideline presents evidence-based recommendations for the diagnostic testing, treatment, and management of KD. Recognizing the comprehensive guidelines developed by the American Heart Association (AHA) for the management of KD (3), the guideline focuses on clinical management questions that are generally posed to rheumatologists, such as the use of adjunctive therapies for initial treatment of severe disease and treatment approaches for refractory disease. Although this guideline may inform an international audience, these recommendations were developed considering experience with and availability of treatment and diagnostic options in the US.
This guideline followed the ACR guideline development process (https://www.rheumatology.org/Practice-Quality/ClinicalSupport/Clinical-Practice-Guidelines) using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) methodology to rate the quality of evidence and develop recommendations (8,9). ACR policy guided the management of conflicts of interest and disclosures (https://www. rheumatology.org/Practice-Quality/Clinical-Support/ClinicalPractice-Guidelines/Vasculitis). Supplementary Appendix 1 (available on the Arthritis & Rheumatology website at http:// onlinelibrary.wiley.com/doi/10.1002/art.42041/abstract) presents a detailed description of the methods.
Sixteen clinical questions addressing the diagnostic testing, treatment, and management of KD were developed by the Core Team, initial Voting Panel, and Expert Panel in the Patient/Population, Intervention, Comparison, and Outcomes (PICO) format. The Literature Review Team undertook systematic literature reviews for prespecified PICO questions. The Expert Panel, including rheumatologists and a pediatric cardiologist, provided expert knowledge to inform discussion of the PICO questions and findings of the literature review. This study did not involve human subjects, and therefore, approval from Human Studies Committees was not required.
The initial Voting Panel comprised 9 adult rheumatologists (SAC, AMA, DLC, PCG, PAM, RLR, PS, JHS, AW), 5 pediatric rheumatologists (LFI, SK, SS, MFI, RPS), and 2 patients (KAF, OIV). They reviewed the Literature Review Team’s evidence summaries and formulated and voted on an initial set of recommendations. To incorporate broader expertise from the pediatric community and validate the results from the initial Voting Panel, a second Voting Panel was established and comprised 8 pediatric rheumatologists new to the Voting Panel (MG, KA, BAB, KH, SKL, MBS, HVM, CY-T), 4 pediatric rheumatologists who participated in both Voting Panels (LFI, SK, SS, RPS), a pediatric infectious disease physician with extensive expertise in KD (AHT), and a pediatric cardiologist with extensive expertise in KD (KF). The second Voting Panel also reviewed the Literature Review Team’s findings and independently formulated and voted on recommendations. Members of the second Voting Panel who were not on the initial Voting Panel were not provided with the recommendations formulated by the initial Voting Panel prior to the second panel’s voting. Each recommendation required consensus from at least 70% of the Voting Panel.
How to interpret the recommendations
A strong recommendation is typically supported by moderate- to high-quality evidence (e.g., multiple randomized controlled trials). For a strong recommendation, the recommended course of action would apply to all or almost all patients. Only a small proportion of clinicians/patients would not want to follow the recommendation. For example, an intervention may be strongly recommended if it is considered low-cost, without harms, and the consequence of not performing the intervention may be catastrophic. An intervention may be strongly recommended against if there is high certainty that the intervention will lead to more harm than the comparison with very low or low certainty about its benefit (10).
A conditional recommendation is generally supported by lower-quality evidence or a close balance between desirable and undesirable outcomes. For a conditional recommendation, the recommended course of action would apply to the majority of patients, but the alternative is a reasonable consideration. Conditional recommendations always warrant a shared decisionmaking approach. We specify conditions under which the alternative may be considered.
In one instance, the committee found that the evidence for a particular PICO question did not support a graded recommendation, and the Voting Panel did not favor one intervention over another. However, the Voting Panel believed the PICO question addressed a commonly encountered clinical question, and thus felt that providing guidance for this question was warranted. For this situation, we present an “ungraded position statement” which reflects general views of the Voting Panel.
In this evidence-based guideline, we explicitly used the best evidence available and present it in a transparent manner for the clinician reader/user (10). In some instances, this includes randomized trials in which the interventions under consideration are directly compared. The GRADE system rates evidence that comes exclusively from the collective experience of the Voting Panel and Patient Panel members as “very low–quality” evidence.
For each recommendation, details regarding the PICO questions and the GRADE evidence tables can be found in Supplementary Appendix 2 (http://onlinelibrary.wiley.com/doi/10.1002/ art.42041/abstract).
This is the first guideline issued by the ACR in conjunction with the Vasculitis Foundation for the management of KD. These recommendations constitute a guide to help physicians treat KD. This guideline should not be used by any agency to restrict access to therapy or require that certain therapies must be utilized prior to other therapies.
Patients with KD in the US are often diagnosed and treated by physicians with expertise in areas other than rheumatology (e.g., pediatric cardiologists, hospitalists, or infectious disease specialists). The recommendations in this guideline focus on situations in which rheumatologists are often consulted, such as in the management of severe disease, treatment-refractory disease, arthritis, and MAS with KD. However, these recommendations are relevant to physicians treating KD regardless of their area of expertise.
The AHA has presented recommendations for the initial treatment and long-term management of KD (3). Recommendations presented in this guideline are intended to supplement the AHA guidelines and serve as an additional reference for rheumatologists who may be less familiar with KD. Emerging evidence suggesting that adjunctive therapy decreases the incidence of severe coronary artery aneurysms and accelerates regression of these aneurysms provides the basis for the recommendations concerning “high-risk” KD (23). Although aneurysms that have regressed are affected by endothelial dysfunction and therefore remain abnormal vasculature, normalized vessel caliber decreases the risk of morbidity from vessel thrombosis (3).
Finally, during the COVID-19 pandemic, a novel multisystem inflammatory syndrome in children (MIS-C) associated with SARS–CoV-2 infection emerged, with some features suggestive of KD (60,61). While as many as 50% of these patients could meet criteria for KD, many patients exhibit manifestations that are unusual for KD, including colitis, myocarditis, and neurologic changes, and often present with or develop shock (62). Further study is needed to understand the relationship between MIS-C and KD, especially Kawasaki shock syndrome. Based on clinical experience, recognition and differentiation of these patients from patients with classic KD is important. Patients who fulfill criteria for KD should be treated using the therapies discussed in this guideline. Additional research is needed to determine optimal treatment for MIS-C with and without KD features.
In summary, the ACR and Vasculitis Foundation present these recommendations to assist physicians in managing KD. This guideline can serve as a resource for basic principles of management and will evolve as new treatment strategies for this disease are identified.
We thank Anne M. Ferris, MBBS, Ora Gewurz-Singer, MD, Rula Hajj-Ali, MD, Eric Matteson, MD, MPH, Robert F. Spiera, MD, Linda Wagner-Weiner, MD, MS, and Kenneth J. Warrington, MD, for serving on the Expert Panel. We thank Antoine G. Sreih, MD and Gary S. Hoffman, MD, MS, for their contributions during the early phases of this project as members of the Core Team. Dr. Hoffman’s participation ended July 2018 due to personal reasons. Dr. Sreih’s involvement ended in December 2018 when he became primarily employed by industry, which precluded his continued participation in this project. We thank Joyce Kullman (Vasculitis Foundation) for her assistance with recruitment for the Patient Panel. We thank the patients who (along with authors Kathy A. Full and Omar I. Vitobaldi) participated in the Patient Panel meeting: Jane Ascroft, Scott A. Brunton, Dedra DeMarco, Thomas Fitzpatrick, Jenn Gordon, Maria S. Mckay, Sandra Nye, Stephanie Sakson, and Ben Wilson. We thank Robin Arnold, Catherine E. Najem, MD, MSCE, and Amit Aakash Shah, MD, MPH, for their assistance with the literature review. We thank the ACR staff, including Ms. Regina Parker, for assistance in organizing the face-to-face meeting and coordinating the administrative aspects of the project, and Ms. Cindy Force for assistance in manuscript preparation. We thank Ms. Janet Waters for help in developing the literature search strategy and performing the initial literature search, and Ms. Janet Joyce for performing the update searches.
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Chung had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Gorelik, Chung, Friedman, Sule, Yildirim-Toruner, Langford, Maz, Abril, Guyatt, Full, Stone, Sundel, Vitobaldi, Husainat, Turgunbaev, Turner, Mustafa.
Acquisition of data. Gorelik, Chung, Binstadt, Lapidus, Kim, Sule, Yildirim-Toruner, Full, Grayson, Stone, Sundel, Byram, Dua, Husainat, James, Kalot, Lin, Springer, Turgunbaev, Villa-Forte, Mustafa.
Analysis and interpretation of data. Gorelik, Chung, Ardalan, Binstadt, Hayward, Imundo, Lapidus, Kim, Son, Sule, Tremoulet, Van Mater, Yildirim-Toruner, Abril, Archer, Conn, Full, Grayson, Ibarra, Merkel, Rhee, Seo, Stone, Sundel, Warner, Byram, Dua, Husainat, Kalot, Lin, Springer, Turgunbaev, Villa-Forte, Mustafa.
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum 2013;65:1–11.
2. Lin MT, Wu MH. The global epidemiology of Kawasaki disease: review and future perspectives. Glob Cardiol Sci Pract 2017;2017:e201720.
3. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation 2017;135:e927–99.
4. Orenstein JM, Shulman ST, Fox LM, Baker SC, Takahashi M, Bhatti TR, et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS One 2012;7:e38998.
5. Burns JC, Kushner HI, Bastian JF, Shike H, Shimizu C, Matsubara T, et al. Kawasaki disease: a brief history. Pediatrics 2000;106:E27.
6. Yanagawa H, Nakamura Y, Yashiro M, Ojima T, Tanihara S, Oki I, et al. Results of the nationwide epidemiologic survey of Kawasaki disease in 1995 and 1996 in Japan. Pediatrics 1998;102:E65.
7. Salgado AP, Ashouri N, Berry EK, Sun X, Jain S, Burns JC, et al. High risk of coronary artery aneurysms in infants younger than 6 months of age with Kawasaki disease. J Pediatr 2017;185:112–6.
8. Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, AlonsoCoello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924–6.
9. Andrews J, Guyatt G, Oxman AD, Alderson P, Dahm P, Falck-Ytter Y, et al. GRADE guidelines: 14. Going from evidence to recommendations: the significance and presentation of recommendations. J Clin Epidemiol 2013;66:719–25.
10. Alexander PE, Gionfriddo MR, Li SA, Bero L, Stoltzfus RJ, Neumann I,et al. A number of factors explain why WHO guideline developers make strong recommendations inconsistent with GRADE guidance. J Clin Epidemiol 2016;70:111–22.
11. Newburger JW, Takahashi M, Burns JC, Beiser AS, Chung KJ, Duffy CE, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med 1986;315:341–7.
12. Kato H, Koike S, Yokoyama T. Kawasaki disease: effect of treatmenton coronary artery involvement. Pediatrics 1979;63:175–9.
13. Newburger JW, Takahashi M, Beiser AS, Burns JC, Bastian J, Chung KJ, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med 1991;324:1633–9.
14. Lo MS, Newburger JW. Role of intravenous immunoglobulin in thetreatment of Kawasaki disease. Int J Rheum Dis 2018;21:64–9.
15. Fabi M, Andreozzi L, Corinaldesi E, Bodnar T, Lami F, Cicero C, et al.Inability of Asian risk scoring systems to predict intravenous immunoglobulin resistance and coronary lesions in Kawasaki disease in an Italian cohort. Eur J Pediatr 2019;178:315–22.
16. Kobayashi T, Saji T, Otani T, Takeuchi K, Nakamura T, Arakawa H,et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet 2012;379:1613–20.
17. Tremoulet AH, Best BM, Song S, Wang S, Corinaldesi E,
Eichenfield JR, et al. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr 2008;153:117–21.
18. Son MB, Gauvreau K, Tremoulet AH, Lo M, Baker AL, de Ferranti S,et al. Risk model development and validation for prediction of coronary artery aneurysms in Kawasaki disease in a North American population. J Am Heart Assoc 2019;8:e011319.
19. Manlhiot C, Yeung RS, Clarizia NA, Chahal N, McCrindle BW. Kawasaki disease at the extremes of the age spectrum. Pediatrics 2009; 124:e410–5.
20. Kato H, Sugimura T, Akagi T, Sato N, Hashino K, Maeno Y, et al.Long-term consequences of Kawasaki disease: a 10- to 21-year follow-up study of 594 patients. Circulation 1996;94:1379–85.
21. Friedman KG, Gauvreau K, Hamaoka-Okamoto A, Tang A, Berry E,Tremoulet AH, et al. Coronary artery aneurysms in Kawasaki disease: risk factors for progressive disease and adverse cardiac events in the US population. J Am Heart Assoc 2016;5:e003289.
22. Miura M, Kobayashi T, Kaneko T, Ayusawa M, Fukazawa R,
Fukushima N, et al. Association of severity of coronary artery aneurysms in patients with Kawasaki disease and risk of later coronary events. JAMA Pediatr 2018;172:e180030.
23. Dionne A, Burns JC, Dahdah N, Tremoulet AH, Gauvreau K, de
Ferranti SD, et al. Treatment intensification in patients with Kawasaki disease and coronary aneurysm at diagnosis. Pediatrics 2019;143: e20183341.
24. Friedman KG, Gauvreau K, Baker A, Son MB, Sundel R, Dionne A,et al. Primary adjunctive corticosteroid therapy is associated with improved outcomes for patients with Kawasaki disease with coronary artery aneurysms at diagnosis. Arch Dis Child 2021;106:247–52.
25. Son MB, Gauvreau K, Kim S, Tang A, Dedeoglu F, Fulton DR, et al.Predicting coronary artery aneurysms in Kawasaki disease at a North American center: an assessment of baseline z scores. J Am Heart Assoc 2017;6:e005378.
26. Dominguez SR, Anderson MS, El-Adawy M, Glodé MP. Preventingcoronary artery abnormalities: a need for earlier diagnosis and treatment of Kawasaki disease. Pediatr Infect Dis J 2012;31:1217–20.
27. McCrindle BW, Li JS, Minich LL, Colan SD, Atz AM, Takahashi M,et al. Coronary artery involvement in children with Kawasaki disease: risk factors from analysis of serial normalized measurements. Circulation 2007;116:174–9.
28. Hamada H, Suzuki H, Onouchi Y, Ebata R, Terai M, Fuse S, et al. Efficacy of primary treatment with immunoglobulin plus ciclosporin for prevention of coronary artery abnormalities in patients with Kawasaki disease predicted to be at increased risk of non-response to intravenous immunoglobulin (KAICA): a randomised controlled, open-label, blinded-endpoints, phase 3 trial. Lancet 2019;393:1128–37.
29. Tremoulet AH, Jain S, Jaggi P, Jiminez-Fernandez S, Pancheri JM,Sun X, et al. Infliximab for intensification of primary therapy for Kawasaki disease: a phase 3 randomised, double-blind, placebocontrolled trial. Lancet 2014;383:1731–8.
30. Yellen ES, Gauvreau K, Takahashi M, Burns JC, Shulman S, Baker AL, et al. Performance of 2004 American Heart Association recommendations for treatment of Kawasaki disease. Pediatrics 2010; 125:E234–41.
31. Burns JC. Frequently asked questions regarding treatment of Kawasaki disease. Glob Cardiol Sci Pract 2017;2017:e201730.
32. Muta H, Ishii M, Yashiro M, Uehara R, Nakamura Y. Late intravenousimmunoglobulin treatment in patients with Kawasaki disease. Pediatrics 2012;129:e291–7.
33. Hu YC, Liu HM, Lin MT, Chen CA, Chiu SN, Lu CW, et al. Outcomes ofKawasaki disease children with spontaneous defervescence within 10 days. Front Pediatr 2019;7:158.
34. Latino GA, Manlhiot C, Yeung RS, Chahal N, McCrindle BW. Macrophage activation syndrome in the acute phase of Kawasaki disease. J Pediatr Hematol Oncol 2010;32:527–31.
35. Wang W, Gong F, Zhu W, Fu S, Zhang Q. Macrophage activation syndrome in Kawasaki disease: more common than we thought?
[review]. Semin Arthritis Rheum 2015;44:405–10.
36. Fukazawa R, Kobayashi T, Mikami M, Saji T, Hamaoka K, Kato H,et al. Nationwide survey of patients with giant coronary aneurysm secondary to Kawasaki disease 1999-2010 in Japan. Circ J 2017;82: 239–46.
37. Kobayashi T, Kobayashi T, Morikawa A, Ikeda K, Seki M, Shimoyawa S, et al. Efficacy of intravenous immunoglobulin combined with prednisolone following resistance to initial intravenous immunoglobulin treatment of acute Kawasaki disease. J Pediatr 2013;163:521–6.
38. Furukawa T, Kishiro M, Akimoto K, Nagata S, Shimizu T, Yamashiro Y. Effects of steroid pulse therapy on immunoglobulinresistant Kawasaki disease. Arch Dis Child 2008;93:142–6.
39. Teraguchi M, Ogino H, Yoshimura K, Taniuchi S, Kino M, Okazaki H,et al. Steroid pulse therapy for children with intravenous immunoglobulin therapy-resistant Kawasaki disease: a prospective study. Pediatr Cardiol 2013;34:959–63.
40. Kim HJ, Lee HE, Yu JW, Kil HR. Clinical outcome of patients withrefractory Kawasaki disease based on treatment modalities. Korean J Pediatr 2016;59:328–34.
41. Sundel RP, Burns JC, Baker A, Beiser AS, Newburger JW. Gammaglobulin re-treatment in Kawasaki disease. J Pediatr 1993; 123: 657–9.
42. Wang Z, Chen F, Wang Y, Li W, Xie X, Liu P, et al. Methylprednisolonepulse therapy or additional IVIG for patients with IVIG-resistant Kawasaki disease. J Immunol Res 2020;2020:4175821.
43. Miura M, Tamame T, Naganuma T, Chinen S, Matsuoka M, Ohki H,et al. Steroid pulse therapy for Kawasaki disease unresponsive to additional immunoglobulin therapy. Paediatr Child Health 2011;16: 479–84.
44. Chan H, Chi H, You H, Wang M, Zhang G, Yang H, et al. Indirectcomparison meta-analysis of treatment options for patients with refractory Kawasaki disease [review]. BMC Pediatr 2019;19:158.
45. Bruggeman CW, Nagelkerke SQ, Lau W, Manlhiot C, de Haas M, vanBruggen R, et al. Treatment-associated hemolysis in Kawasaki disease: association with blood-group antibody titers in IVIG products. Blood Advances 2020;4:3416–26.
46. Roberts SC, Jain S, Tremoulet AH, Kim KK, Burns JC, KIDCARE Multicenter Study Group, et al. The Kawasaki Disease Comparative Effectiveness (KIDCARE) trial: a phase III, randomized trial of second intravenous immunoglobulin versus infliximab for resistant Kawasaki disease. Contemp Clin Trials 2019;79:98–103.
47. Burns JC, Roberts SC, Tremoulet AH, He F, Printz BF, Ashouri N,et al. Infliximab versus second intravenous immunoglobulin for treatment of resistant Kawasaki disease (KIDCARE): a randomized comparative effectiveness trial. Lancet Child Adolesc Health 2021;5: 852–61.
48. Pendergrast J, Armali C, Callum J, Cserti-Gazdewich C, Jiwajee A,Lieberman L, et al. A prospective observational study of the incidence, natural history, and risk factors for intravenous immunoglobulin-mediated hemolysis. Transfusion 2021;61:1053–3.
49. Kim GB, Yu JJ, Yoon KL, Jeong SI, Song YH, Han JW, et al. Mediumor higher-dose acetylsalicylic acid for acute Kawasaki disease and patient outcomes. J Pediatr 2017;184:125–9.
50. Dallaire F, Fortier-Morissettee Z, Blais S, Dhanrajani A, Basodan D,Renaud C, et al. Aspirin dose and prevention of coronary abnormalities in Kawasaki disease. Pediatrics 2017;139:e20170098.
51. Amarilyo G, Koren Y, Simon DB, Bar-Meir M, Bahat H, Helou MH,et al. High-dose aspirin for Kawasaki disease: outdated myth or effective aid? Clin Exp Rheumatol 2017;35 Suppl:209–12.
52. Koren G, Lavi S, Rose V, Rowe R. Kawasaki disease: review of riskfactors for coronary aneurysms. J Pediatr 1986;108:388–92.
53. Lee KY, Oh JH, Han JW, Lee JS, Lee BC. Arthritis in Kawasaki disease after responding to intravenous immunoglobulin treatment. Eur J Pediatr 2005;164:451–2.
54. Gong GW, McCrindle BW, Ching JC, Yeung RS. Arthritis presentingduring the acute phase of Kawasaki disease. J Pediatr 2006;148: 800–5.
55. Martins A, Conde M, Brito M, Gouveia C. Arthritis in Kawasaki disease: a poorly recognised manifestation. J Paediatr Child Health 2018;54:1371–4.
56. Antman EM. The aspirin-NSAID interaction: more data, but a lack ofclarity remains. J Am Coll Cardiol 2018;71:1752–4.
57. Sonobe T, Kiyosawa N, Tsuchiya K, Aso S, Imada Y, Imai Y, et al.Prevalence of coronary artery abnormality in incomplete Kawasaki disease. Pediatr Int 2007;49:421–6.
58. Kanegaye JT, Wilder MS, Molkara D, Frazer JR, Pancheri J, Tremoulet AH, et al. Recognition of a Kawasaki disease shock syndrome. Pediatrics 2009;123:e783–9.
59. Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: diagnosis and management. Paediatr Drugs 2020; 22:29–44.
60. Whittaker E, Bamford A, Kenny J, Kaforou M, Jones CE, Shah P, et al.Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA 2020;324:259–69.
61. Kaushik S, Aydin SI, Derespina K, Bansal PB, Kowalsky S, Trachtman R, et al. Multisystem inflammatory syndrome in children (MIS-C) associated with SARS-CoV-2 infection: a multi-institutional study from New York City. J Pediatr 2020;224:24–9.
62. Cheung EW, Zachariah P, Gorelik M, Boneparth A, Kernie SG, Orange JS, et al. Multisystem inflammatory syndrome related to COVID-19 in previously healthy children and adolescents in New York City. JAMA 2020;324:294–6.
Table 1 presents definitions of selected terms used in the recommendations, and Table 2 presents the recommendations with their supporting PICO questions and level of evidence. Figure 1 shows a schematic highlighting the key recommendations for the treatment of KD. Conditional recommendations are indicated as such and are generally conditional due to a lack of high-quality evidence (e.g., multiple randomized controlled trials) supporting the recommendation.
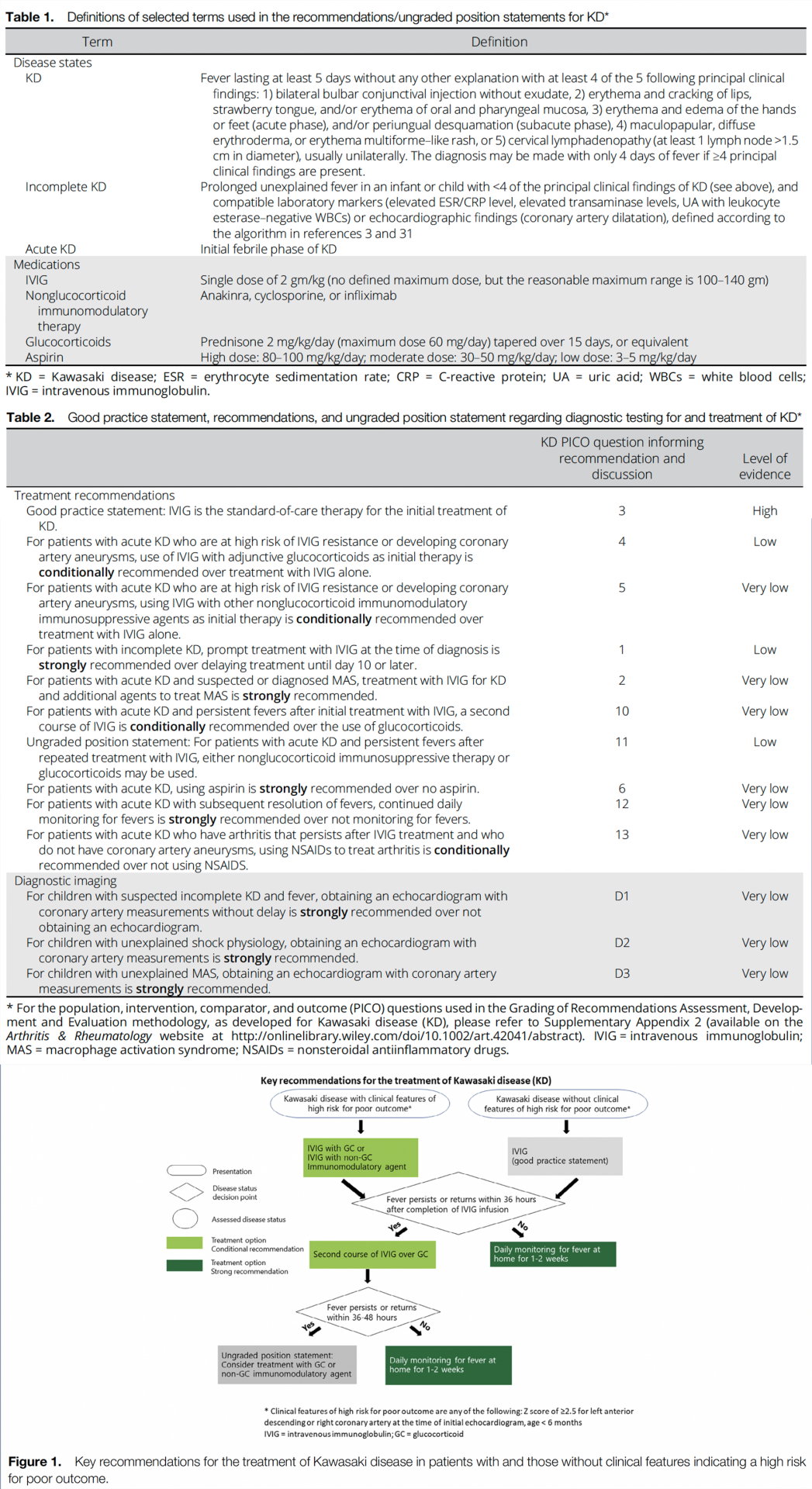
Good Practice Statement: IVIG is the standard-of-care therapy for the initial treatment of KD.
IVIG has been established as the standard-of-care treatment for KD for the last 4 decades due to the significant reduction in the rate of coronary artery aneurysms as well as a reduction in the duration of fever and other symptoms associated with its use (11). Although no comparative trials of IVIG and glucocorticoids have been conducted, glucocorticoids alone were considered inadequate to treat KD based on findings from an early case series (12,13). Thus, IVIG use is a mainstay of KD therapy. The mechanisms of the therapeutic effect of IVIG in KD are still under study (14).
Recommendation: For patients with acute KD who are at high risk of IVIG resistance or developing coronary artery aneurysms, use of IVIG with adjunctive glucocorticoids as initial therapy is conditionally recommended over treatment with IVIG alone.
Risk scores have been developed to identify patients at high risk of IVIG resistance in Japanese populations, but these scores had poor sensitivity and specificity in a multiethnic US population (15–17). A recent study of patients in North America identified the following demographic and clinical characteristics as predictors for the development of coronary artery aneurysms at 2–8 weeks: a Z score in the left anterior descending or right coronary artery of ≥2.0, age <6 months, Asian race, and a C-reactive protein level of ≥13 mg/dl (18). In the US, features that suggest an increased likelihood of morbidity in KD include age <6 months or >9 years at diagnosis (19,20). In patients with giant aneurysms (Z score >10) or multiple aneurysms, regression of the aneurysms (via remodeling) at late stages (21) is less likely, and large aneurysms are associated with the greatest morbidity rate (21,22).
Emerging evidence suggests that adding glucocorticoids to IVIG as primary therapy can decrease the risk of developing coronary artery aneurysms, although the strongest evidence is from a Japanese population (16). There is also emerging evidence that the addition of glucocorticoids can decrease the progression of coronary artery aneurysms in patients with coronary aneurysms at the time of diagnosis (23,24).
Thus, the use of glucocorticoids with IVIG is a treatment option in patients who are at high risk of coronary artery aneurysms. For this recommendation, the Voting Panel defined high-risk features as a Z score of ≥2.5 (25–27) for the left anterior descending or right coronary artery at the time of the initial echocardiography and age <6 months. This definition uses the Z score of 2.5 instead of 2.0, since a score of 2.5 is defined as representing a true aneurysm. We also do not include patients >9 years of age, as only a portion of these patients are believed to be “high-risk.” The Voting Panel acknowledges that further investigation clarifying the features of “high-risk” KD in a US population is needed. Optimal dosing and duration of glucocorticoids are still to be determined and will require further study in a US population, but a typical dosage would be prednisone starting at 2 mg/kg/day (maximum 60 mg/day), with the dose tapered over 15 days. The use of IVIG alone is conditionally recommended when the treating physician is unsure whether a patient is at high risk of developing coronary artery aneurysms, or when additional glucocorticoid exposure may be detrimental to the patient. The use of glucocorticoids is generally not recommended in patients who do not have features suggestive of high risk.
Recommendation: For patients with acute KD who are at high risk of IVIG resistance or developing coronary artery aneurysms, using IVIG with other nonglucocorticoid immunomodulatory immunosuppressive agents as initial therapy is conditionally recommended over treatment with IVIG alone.
For patients with features of high-risk disease, adjunctive therapy with a nonglucocorticoid immunomodulatory agent, such as infliximab, anakinra, or cyclosporine, may improve outcomes in acute disease and may also improve cardiac outcomes (21,23,28,29). Thus, adjunctive therapy with a nonglucocorticoid immunomodulatory agent can be considered in patients with KD who are at high risk of not responding to IVIG. For any patient considered to have features of high-risk disease, a rheumatologist or other clinician with expertise in KD should be consulted before adding an adjunctive agent (glucocorticoids or nonglucocorticoid immunomodulatory agents) to ensure correct diagnosis and appropriate utilization of adjunctive therapies. The Voting Panel also advocated for a stepwise therapeutic algorithm, in which either a glucocorticoid or nonglucocorticoid immunosuppressive agent along with IVIG is initially prescribed in high-risk patients. Currently, there is more evidence supporting the use of glucocorticoids than nonglucocorticoid immunosuppressive agents in this patient population, and further study is warranted to compare the efficacy of glucocorticoid therapy to nonglucocorticoid therapy in this population. There may be specific situations in which glucocorticoids are contraindicated, but the patient exhibits high-risk features that would indicate the need for adjunctive nonglucocorticoid immunomodulatory therapy. Once again, use of IVIG alone is appropriate if the treating physician is unsure whether a patient has high-risk features, as described in the previous recommendation, or if using a nonglucocorticoid immunomodulatory agent presents a greater risk of an adverse effect to the patient.
Recommendation: For patients with incomplete KD, prompt treatment with IVIG at the time of diagnosis is strongly recommended over delaying treatment until day 10 or later.
Incomplete KD is defined as a condition of suspected KD that lacks the sufficient number of features to meet criteria for KD (3,30) (Table 1). Patients with incomplete KD should be evaluated using the algorithm for incomplete KD as outlined in the AHA guidelines for KD (3,30). The performance of this algorithm for identifying patients with incomplete KD has been validated (30). The practice of completing diagnostic testing and initiating treatment by day 10 of fever is not based on mechanisms or studies of disease pathogenesis or treatment outcomes. Instead, this “treatment deadline” was chosen as a research end point in the original studies in KD (31). It is recognized that prompt treatment prevents adverse outcomes, and patients should receive treatment as soon as the diagnosis of incomplete KD is made, rather than waiting until day 10 to see if they meet criteria for complete KD. Although delayed IVIG treatment after 10 days of fever is associated with worse outcomes (32), IVIG treatment should still be administered to patients even if they present after >10 days of fever. Thus, patients who meet the criteria for incomplete KD according to the AHA guidelines should be treated promptly to avoid adverse consequences, since the risk of developing coronary artery dilatation increases each day without treatment. This is a strong recommendation because this intervention is standard of care, and the consequence of delaying treatment may lead to significant coronary artery aneurysms or rupture, which are severe adverse outcomes. In addition, resolution of fever prior to day 10 is not an indication to withhold treatment in patients meeting criteria for incomplete or complete KD. In patients with characteristically elevated levels of acute-phase reactants, treatment is recommended, as these patients remain at high risk of developing adverse outcomes (3,33).
Recommendation: For patients with acute KD and suspected or diagnosed macrophage activation syndrome (MAS), treatment with IVIG for KD and additional agents to treat MAS is strongly recommended.
MAS, a form of secondary hemophagocytic lymphohistiocytosis (HLH), is a potentially underrecognized complication of KD (34,35). Formal diagnostic criteria for MAS in the setting of KD have not been developed. However, drawing on experience with other secondary HLH presentations, MAS may be suspected in KD patients with persistent fever, splenomegaly, elevated ferritin levels, and thrombocytopenia (35). Inadequate treatment of either KD or MAS could result in severe consequences. These include large coronary aneurysms or coronary artery stenosis, leading to death via cardiac infarct or coronary rupture in KD, or death due to multiorgan dysfunction in MAS (36).
Thus, to ensure appropriate therapy, each disease entity should be considered separately with appropriate targeted therapy. KD should be treated with IVIG as the first-line therapy, and MAS should also be treated with appropriate agents for targeting cytokine storm or underlying triggers. Anakinra and glucocorticoids are preferred for treatment in these patients over a primary HLH–directed treatment protocol with cytotoxic agents. Although no head-to-head comparison trials have been published, primary HLH–directed therapy, which has considerably more associated toxicity, may not be warranted in patients without an underlying genetic predisposition to HLH. This is a strong recommendation because the consequence of not appropriately treating KD or MAS may be associated with increased mortality.
Recommendation: For patients with acute KD and persistent fevers after initial treatment with IVIG, a second course of IVIG is conditionally recommended over the use of glucocorticoids.
Findings from 6 studies provide indirect evidence for this recommendation, though no direct comparative studies were available (37–42). For KD patients with persistent fevers after the initial course of IVIG, studies suggest that there is no difference in coronary artery outcomes between repeating the course of IVIG versus a single dose of pulse glucocorticoids (i.e., 30 mg/kg with a maximum of 1 gm) (43). A meta-analysis of 12 studies involving 372 patients whose disease was resistant to treatment with a single course of IVIG failed to demonstrate significant differences in coronary artery outcomes between patients treated with a second dose of IVIG versus glucocorticoids versus infliximab (44). Although current evidence does not clearly indicate the superiority of a second course of IVIG over glucocorticoids, a second course of IVIG in patients who have persistent fever for >36 hours after the first dose is conditionally recommended, as it is the current standard of care. However, as a conditional recommendation, glucocorticoids are a reasonable alternative (e.g., starting at 2 mg/kg/day and tapering over 15 days or a single dose of 20–30 mg/kg). Repeated doses of IVIG may put patients at risk of hemolytic anemia, and the results of ongoing studies may alter this recommendation in the future (45–47). In patients with risk factors for hemolytic anemia with IVIG, such as non–type O blood groups, alternative therapies, such as glucocorticoids or nonglucocorticoid immunomodulatory therapy, should be considered (45,48). Combination therapy (i.e., multiple anticytokine agents) is not recommended for routine care and generally should only be considered in patients with extremely severe disease.
Ungraded Position Statement: For patients with acute KD and persistent fevers after repeated treatment with IVIG, either nonglucocorticoid immunosuppressive therapy or glucocorticoids may be used.
In patients with KD and persistent fevers after 2 doses of IVIG, use of another agent is indicated. Findings from studies of infliximab and cyclosporine for the treatment of refractory KD suggest some potential benefit of these agents (28,29). Head-tohead comparisons between these nonglucocorticoid immunosuppressive agents and glucocorticoids have not been performed. There is no clinical evidence to suggest superiority of either glucocorticoids or nonglucocorticoid immunomodulatory agents, and the Voting Panel was evenly split as to which to recommend. Thus, the Voting Panel believes that use of either class of agents would be appropriate based on the specific clinical scenario. Combination therapy (glucocorticoid with a nonglucocorticoid immunosuppressive agent) can be considered in severe cases, such as rapidly expanding aneurysms or imminently lifethreatening disease.
Recommendation: For patients with acute KD, using aspirin is strongly recommended over no aspirin.
The use of aspirin in patients with KD to reduce inflammation and prevent thrombosis through its antiplatelet effect is considered standard of care. However, the optimal dosage is unclear.
Historically, high dosages of aspirin (80–100 mg/kg/day) were used during the acute phase for antiinflammatory effects, but there is no evidence of benefit with high- versus low-dose aspirin (3–5 mg/kg/day) when considering coronary vascular damage (49–51). The AHA guidelines should be consulted for recommendations regarding anticoagulation in patients with larger aneurysms (3). This is a strong recommendation because aspirin is recognized to be a low-cost intervention with limited toxicity at antiplatelet dosing, and also because there are significant consequences of not inhibiting platelet activity, such as coronary artery thrombosis.
Recommendation: For patients with acute KD with subsequent resolution of fevers, continued daily monitoring is strongly recommended over not monitoring for fevers.
Patients with KD may experience recurrence of disease or treatment-refractory disease heralded by returning fever and other symptoms. In addition, the duration of fever is a predictor of coronary artery aneurysms (52). Thus, patients should be monitored daily for fevers for 1–2 weeks after discharge, with fever defined as an oral temperature in older children and a rectal temperature in infants of >38.0C or an axillary temperature of >37.5C. Parents or guardians should be instructed by the discharging physician on how to take a temperature and told to contact the physician should fever recur. Daily fever monitoring is strongly recommended because it is low cost, without harms, and may identify recurring KD.
Recommendation: For patients with acute KD who have arthritis that persists after IVIG treatment and who do not have coronary artery aneurysms, using nonsteroidal antiinflammatory drugs (NSAIDs) to treat arthritis is conditionally recommended over not using NSAIDS.
Patients with acute and subacute KD can develop arthritis, but treatment with IVIG decreases the incidence of arthritis in this setting from 30% to ~2–12% (53). Arthritis in KD is usually shortlived, lasting between 7 and 21 days (53–55). In general, arthritis can be treated with NSAIDs, but NSAIDs directly inhibit the ability of aspirin to acetylate cyclooxygenase 1, thus decreasing aspirin’s protective effects (56). The impact of NSAID use can be partially mitigated by administering aspirin prior to NSAIDs, but combination therapy is usually not recommended given the potential increased toxicity (56).
Patients without coronary artery aneurysms generally do not require long-term treatment with aspirin. The optimal duration of treatment with aspirin in these patients is not well defined but in practice is generally 6–8 weeks. Patients with coronary artery aneurysms require long-term and potentially indefinite aspirin use (3). For patients without coronary artery aneurysms who do not require long-term use of aspirin and who have arthritis requiring additional therapy, aspirin can be temporarily suspended and a short course (3–4 weeks) of NSAIDs can be used as needed.
Conversely, if indefinite or long-term aspirin use is needed due to coronary artery aneurysms, then either acetaminophen, a short course of glucocorticoids, or nonsystemic NSAID pain management options (e.g., a topical NSAID) can be used. A pediatric hematologist or cardiologist should be consulted to consider an alternative anticoagulant (e.g., clopidogrel) if prolonged use of systemic NSAIDs is required (i.e., >3 weeks), especially in a patient with coronary artery aneurysms.
Recommendation: For children with suspected incomplete KD and fever, obtaining an echocardiogram with coronary artery measurements without delay is strongly recommended over not obtaining an echocardiogram.
Patients with incomplete KD have at least the same or an increased risk of coronary artery lesions compared to patients with classic KD (57). Therefore, in patients in whom incomplete KD is suspected, a diagnosis should be confirmed as soon as possible so that features associated with high risk of coronary artery aneurysms can be assessed, treatment can be initiated without delay, and adverse outcomes can be prevented. Echocardiographic assessment of the absolute dimensions of the coronary artery and body surface area–adjusted Z scores can help establish a diagnosis of incomplete KD and should be obtained promptly when this diagnosis is suspected. The AHA guidelines for KD management provide an algorithm for the evaluation and treatment of suspected incomplete KD (3), which has been validated (30). Echocardiography is strongly recommended in this scenario because it has minimal potential harms and may prevent adverse outcomes by prompting the treatment of incomplete KD.
Recommendation: For children with unexplained shock physiology, obtaining an echocardiogram with coronary artery measurements is strongly recommended.
Unexplained shock can be due to Kawasaki shock syndrome, a recognized manifestation of KD (58). Echocardiograms are often obtained in children with unexplained shock, especially in those with the prodrome of prolonged fever, to evaluate cardiac function and help identify the potential etiology. We recommend that the coronary arteries be included in the echocardiographic examination to evaluate for KD as a potential etiology of shock. This is strongly recommended because echocardiography has minimal potential harms and may prevent catastrophic outcomes by facilitating the diagnosis and prompt treatment of KD.
Recommendation: For children with unexplained MAS, obtaining an echocardiogram with coronary artery measurements is strongly recommended.
As noted above, KD is one of the clinical inflammatory entities that can predispose patients to the development of MAS (59). For patients with unexplained MAS, KD should be considered as a potential underlying etiology. Echocardiography with inclusion of the coronary arteries is one method of identifying KD as a potential treatable underlying condition. This is strongly recommended because echocardiography has minimal potential harms and may prevent adverse outcomes by facilitating the diagnosis and prompt treatment of KD.